Part 1: 505(b)(2) NDA – Navigating the Regulatory Pathway

The 505(b)(2) is a New Drug Application (NDA) containing full reports of safety and effectiveness, where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference for use.
This is in contrast to the stand-alone 505(b)(1) ANDA application, in which all safety and efficacy data required comes from sponsor-conducted studies, as well as the ANDA (generic) application where this data comes entirely from reference to an already-approved formulation. By design, the 505(b)(2) encompasses a wide variety of submission scenarios, ranging from moieties that could almost be approved as generics up to those supported by multiple sponsor-conducted Phase 3 studies.
Sponsors can refer to the FDA’s detailed guidance to determine if their candidate product falls under a 505(b)(2) scenario.
The 505(b)(2) pathway is a subject of intense interest to sponsors who are seeking rapid and cost-effective pathways for drug approval. It can reduce risks relative to the 505(b)(1) while conferring a degree of market exclusivity commensurate with clinical development effort (3, 5, or 7 years, dependent on the conduct of a Phase 2/3 study, new chemical entity status, and orphan status, respectively).
The concept of commensurate effort and benefit is important, since the process offers expediencies, market exclusivity, and challenges in equal measure. Because the amount of data presented within a 505(b)(2) can range anywhere between that of an ANDA and a 505(b)(1), development and planning can be unpredictable and intimidating.
This series reviews the unique challenges of the application type and describes tools sponsors can use to put their best foot forward during the early stages of 505(b)(2) planning and throughout development. Individually these tools are simple, and none are unique to the 505(b)(2). Used proactively and in combination, they become powerful time-savers.
Regulatory History and the Challenge of 505(b)(2) Application Structure
A historically informed approach to the 505(b)(2) builds an appreciation for structural challenges inherent in the application type. Sponsors of currently marketed unapproved drugs may also wish to review the appendix in the FDA Marketed Unapproved Drugs Compliance Policy Guide for additional historical context.
The application pathway came into existence in 1984 as part of the Drug Price Competition and Patent Term Restoration Act, also known as the Hatch-Waxman Act. This act replaced a cumbersome literature-based application process for generics with a legal structure for approval centered on the relationship between the applicant formulation and a previously approved Reference Listed Drug (RLD).
Hatch Waxman supports this structure with a clinical methodology relating applicant formulations and RLDs: bioequivalence studies in the case of ANDAs and relative bioavailability in the case of the 505(b)(2). These concepts form a kind of regulatory Occam’s razor, ensuring a simple and consistent means of relating the labeling claims of generics and 505(b)(2) compounds with RLDs.
The existence of a simple means to relate reference formulations and unapproved drugs does not always result in a simple application. In fact, the complexity of the 505(b)(2) can rival standalone 505(b)(1) NDAs in a couple of key areas: application structure and supportive data for label claims.
To illustrate this, consider the applications for three different products: a reference drug approved through the 505(b)(1) application pathway, an ANDA product, and a 505(b)(2) product (Table 1).
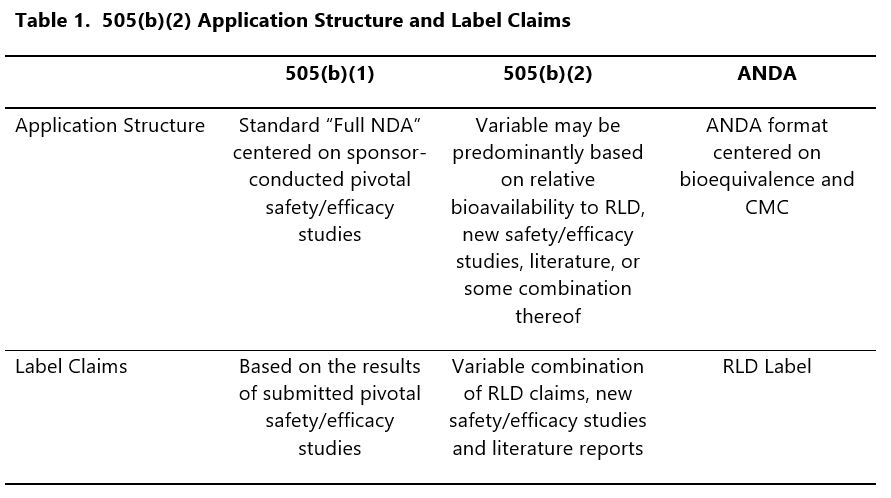
In the case of the 505(b)(1) and ANDA, the relationship between label claims and supportive data is generally well-defined by, and within, the application structure. This is most obvious in the case of the ANDA, where a single bioequivalence study typically results in a label identical to the RLD; it is also generally true of the 505(b)(1), where the organization of the submission relates label claims directly to adequate and well controlled Phase 3 studies.
In contrast, the 505(b)(2) presents a variable scenario where label claims may be derived from the RLD, new studies, or even literature.
Stay tuned for next week’s blog article where we discuss the ongoing task of directly and specifically relating these claims to the application and more.
by Ben Kaspar
About the author
Ben Kaspar is Global Submissions Manager at MMS, based in San Diego, California.
Suggested For You

perspectives
July 30th, 2024
The Critical Role of Quality Control (QC) – Medical Writing and Beyond

perspectives
July 23rd, 2024
PSI 2024 Ignited Conversations on External Data Sources, Requirements for Estimands, and Bayesian Methodology for Statisticians in Pharma

perspectives
July 16th, 2024
Key Steps to Successful CMC Authoring of IND and IMPD Submissions

perspectives
July 9th, 2024
Managing RTOR Submissions: How to Run a Successful Race from the Top Line Starting Line
perspectives
July 2nd, 2024
Part 1: RWD Noninterventional Study Design and FDA Engagement Opportunity for Early Stage Oncology

perspectives
June 21st, 2024
Peer-Reviewed Journal Articles: The Crucial Role of Publication in the Pharmaceutical Industry

perspectives
June 14th, 2024
A Structured Approach to Benefit-Risk Assessment Throughout Product Development in the Pharmaceutical Industry

perspectives
June 6th, 2024
Datacise and Diversity in Patient Enrollment: Combining Geospatial and Demographic Data to Aid Site Selection

perspectives
May 29th, 2024
Confined Deferrals in Clinical Trial Applications: Anticipating the Revised EU CTR Transparency Rules
perspectives
May 21st, 2024
Psychedelics and Regulatory Considerations Part II: A Shift in Lexicon and Implications of “Nonmedical Use” On Labelling

perspectives
May 10th, 2024
Psychedelics in Drug Development and Regulatory Considerations Part I: Benefit-Risk

perspectives
April 29th, 2024
Validation of Clinical Dashboards for Decision Making